26.05.2026 | Published by bit.bio

Biological discovery is moving into a new phase. Ideas that once felt speculative, like virtual cells, or the ability to computationally predict how a cell will respond to a new stimulus, are beginning to materialise. Meanwhile, cell identity, once viewed as the outcome of irreversible developmental processes, has proven to be programmable with increasing precision and scale. At the intersection of these two ideas, we can imagine a near future where cell programming becomes more systematic, more predictive, and more like programming a computer.
Reaching that future will depend on more than just better models. It will also require the right data. Large-scale cell atlases have transformed our ability to catalogue observed cell states, but virtual cell models need to go further by predicting how cellular state changes in response to genetic, chemical or environmental inputs. Training and testing those models requires systematic perturbation datasets that connect defined biological inputs to measurable cellular outcomes. Generating data at this level of scale and consistency remains a challenge, particularly because existing perturbation datasets are often sparse, context-dependent, and difficult to compare across experimental systems, technologies and starting cell states.
Addressing this bottleneck is central to our work at bit.bio. Over the past several years, we have built a cell programming platform designed not only to manufacture defined iPSC-derived human cell products, but also to systematically map how perturbations shape cell identity at industrial scale. Our most important tool for doing this is screening, where we test many cellular perturbations in a single experiment. We run everything from narrow screens, focused on a specific cell type or function, to broad screens designed to capture as many perturbations as possible.
One of our earliest broad screens was designed to comprehensively map the effects of single transcription factor overexpression across multiple contexts. It remains a distinctive dataset because of its scale, the perturbation system behind it, and the breadth of context built into the design. In this post, we introduce the technologies that made it possible and some of the exciting biology that this dataset is allowing us to understand, from interactions between context and perturbation to isoform specific effects.
How we select and control perturbations
If perturbation maps are the route to more predictive models of cell behaviour, transcription factors are an obvious place to start. By binding DNA and regulating gene expression, they can activate lineage-specific programmes, repress alternative fates and reshape the regulatory networks that define cell identity. Cell programming leverages this by using controlled transcription factor expression to shift cells from one state to another. The right combinations of factors can induce the transition from induced pluripotent stem cells (iPSCs) into stable mature cell type identities, but the examples we know today are still needles in a haystack. The wider possibilities of this space remain largely unexplored.
To really understand this branch of synthetic biology, we need to be able to predict how combinations of transcription factors influence cell state. The combinatorics quickly become daunting: around 2,000 single perturbations expand to roughly 2 million pairs and 1.3 billion triples. We cannot test every combination directly, but understanding where combinations behave differently from the sum of their parts can make the problem more approachable. The first step is to cover the single-factor space as broadly as possible. To support that, bit.bio developed the ioLibrary, which comprises 2,312 opti-ox overexpression vectors, each carrying a single transcription factor isoform together with a unique identifying barcode.
To deliver and control these constructs, we used opti-ox, the same inducible overexpression system we use in our programming platform. In opti-ox, engineered transcription factor constructs are inserted into genomic safe harbour sites and activated with doxycycline, giving tight control over both where and when expression occurs. Because expression is driven independently of the endogenous locus, perturbations can be assessed even when the native gene is inaccessible due to chromatin state. This also gives us control over the design of the perturbation itself: by including defined transcription factor isoforms, we can begin to ask not only whether a factor matters, but whether different versions of the same factor produce meaningfully different outcomes.
The barcodes incorporated into each vector add another important layer of information. Because each barcode is expressed along with its construct, targeted sequencing allows us to assess the level of perturbation expression, rather than simply confirming vector delivery. This makes perturbation assignment more direct and distinguishes direct exogenous expression from endogenous activation.
How we sequence
The breadth of possible outcomes in this experiment meant that the sequencing method had to work for any cell type the screen might produce. Parse Bioscience’s split-pool sequencing technology offers several advantages here. Because cell barcoding takes place within intact cells rather than relying on individual cell capture, the workflow can accommodate diverse cell types and states, which is particularly useful in discovery screens where the resulting cell states are not known in advance. It also makes large-scale experiments across many samples and conditions more practical to run, with natural pause points that make the workflow easier to process in stages. Alongside single-cell gene expression profiling, we used a custom multimodal readout with parallel amplicon sequencing of the overexpressed transcription factors, allowing perturbation identity and resulting cell state to be captured within the same experimental framework. For this screen, that combination of scalability, practicality and compatibility with multimodal readout was a major advantage of the split-pool approach.
How we run the screen
The same transcription factor may behave very differently in different cell states and the screen was designed to ask not only what happens when individual transcription factors are overexpressed, but how those effects change across different biological contexts. To introduce that variation in a controlled way, we ran the screen in a single iPSC line across four culture conditions: a neutral, proliferation-biased E6 medium and three germ layer priming media biased toward ectoderm, mesoderm and endoderm.
Cells were transfected with the ioLibrary using opti-ox delivery and then cultured for five days under the following conditions. In the germ layer media, cells spent two days in priming media followed by three days in E6, while cells in the E6 medium remained in E6 throughout the full five-day period. Figure 1 shows how perturbation, context and replicate structure were combined in the overall design of the screen. For each culture condition, three replicates were cultured with doxycycline induction, while one replicate was cultured without doxycycline as a control, giving 16 samples in total. The full experiment was then processed through a unified split-pool workflow on a Parse Evercode Mega kit, targeting around 1 million cells and generating a combined readout of cell state and perturbation identity across the dataset.
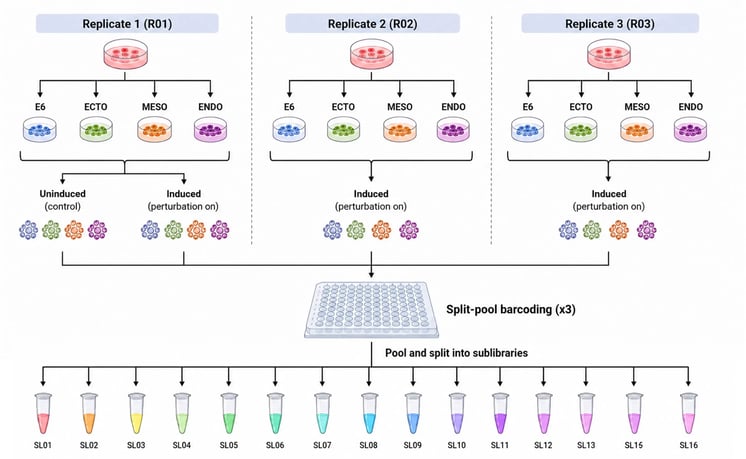
Figure 1. Screen design and sample generation workflow. Three biological replicates were cultured across four media conditions (E6, ectoderm, mesoderm and endoderm). For each replicate, induced and uninduced control samples were generated, combined, and processed through a single Parse Mega Kit split-pool workflow, producing 16 sub-libraries and around 1 million cells overall.
After filtering, the dataset comprised around 712,000 high-quality cells with roughly 590,000 containing at least one overexpressed transcription factor. Assigning perturbations is a delicate process prone to false negative and positive errors in different perturbation contexts. We developed a statistical procedure to weigh expression level against background and confidently assign perturbations while building in experiment context via priors. Across the ioLibrary, 1,443 transcription factors, or 62% of the full library, were detected in at least one cell. The dataset also showed strong overall quality, with a median of 3,930 genes and 7,792 UMIs captured per cell. Across the three biological replicates in Figure 2, we observed strong overlap between cell populations, consistent with the reproducibility of the underlying perturbation system.

Figure 2. Replicate consistency. Across the three replicates we see nearly identical UMAP structure.
What we learn
As expected, dimensionality reduction shows that culture condition is the dominant source of variation in the dataset. In the UMAP, cells separate into broad transcriptional neighbourhoods corresponding to the four culture conditions (Figure 3B), with the uninduced controls occupying regions associated with each condition (Figure 3A’’). This indicates that the media successfully establishes distinct baseline cell states to compare the effects of perturbations.
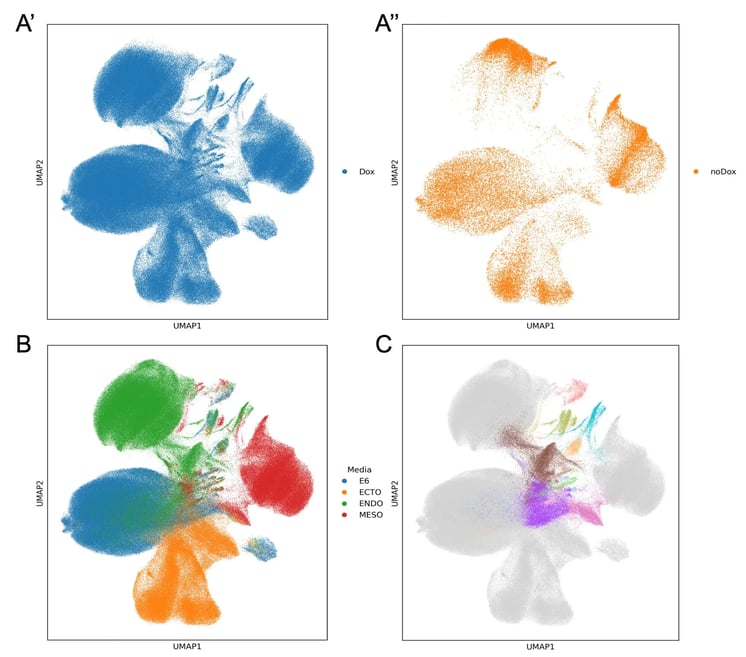
Figure 3. The screen captures induced cell state changes across multiple biological contexts. UMAPs of the QC-filtered dataset showing A’, induced cells; A’’, uninduced controls; B, induced cells coloured by media condition; and C, selected induced subclusters spanning multiple media conditions that were selected for re-clustering.
Within the media-driven cells, we also see clear evidence of perturbations. Several selected clusters contain cells from multiple culture conditions and include few to no uninduced cells (Figure 3C), showing that transcription factor overexpression drives clear shifts in cell state beyond the effects of media. Perturbation induced clusters are not often visible in a UMAP and this shows the effectiveness of opti-ox at driving global transcriptomic changes.
The broad variation from culture condition can obscure differences within these perturbed populations. To better resolve these effects, we focused on the induced clusters that spanned multiple media conditions and reclustered them separately. This reduces the influence of the larger undifferentiated and media-dominated regions of the dataset, allowing transcription factor-associated clusters to be examined at higher resolution.
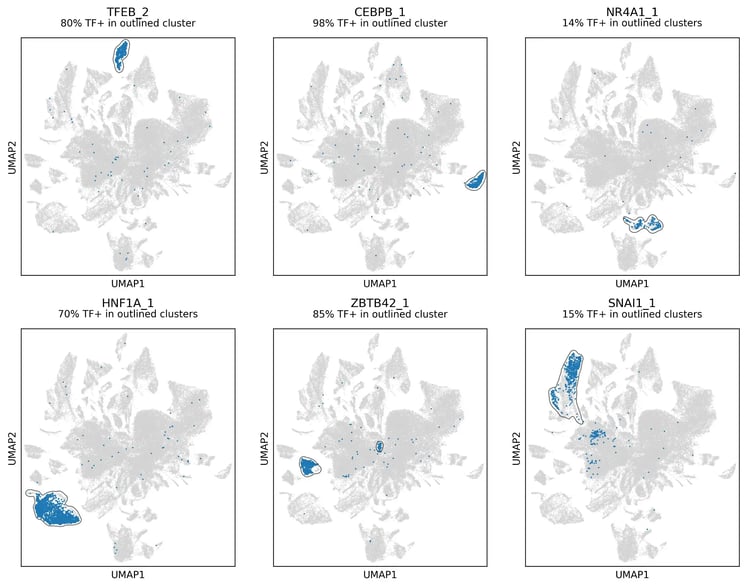
Figure 5. Transcription factor specific clusters. Many clusters are specific to particular transcription factor isoforms, showing that these factors can drive cells toward consistent states. Numerical suffix denotes isoform number.
Having established that the reclustered structure is not simply explained by media condition or cell cycle, we next asked whether it could be linked directly to the overexpressed transcription factors. When we reveal overexpressed transcription factor identity within the reclustered UMAP, specific factors are strongly enriched in distinct peripheral clusters (Figure 5). This suggests that much of the local structure in these regions is being driven by the perturbation itself.
In several cases, cells carrying the same transcription factor converge transcriptionally despite coming from different media conditions. This is important because it suggests that the clustering is not just a general response to induction, current proliferation state or culture condition. Instead, defined transcription factors can drive cells towards reproducible transcriptional outcomes across biological contexts.
The strength of this effect varies between factors. Some transcription factors form compact, well-defined clusters, consistent with strong and reproducible shifts in cell state. Others are more broadly distributed, suggesting more variable effects. This range of behaviours is exactly what this screen was designed to capture: not only whether a transcription factor can perturb cell identity, but how consistently it does so, and how that effect depends on the cellular context in which it is expressed, including the primed state created by the media conditions.
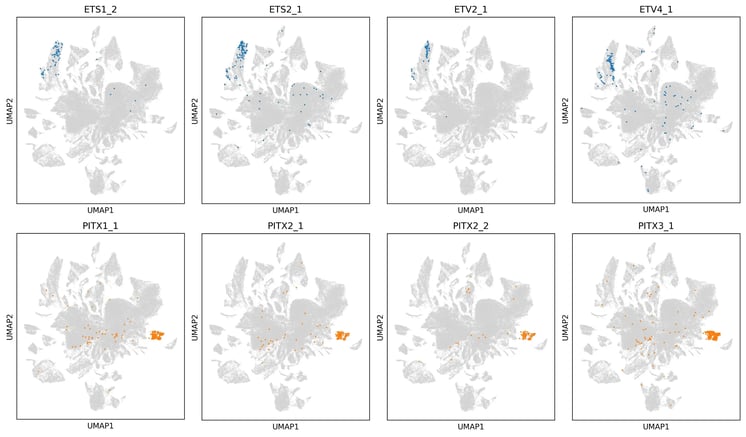
Figure 6. Related transcription factor family members occupy shared regions of transcriptional space. UMAPs show selected ETS family members (top) and PITX family members (bottom) overlaid on the reclustered induced population. Family members are enriched in neighbouring or partially overlapping regions, consistent with shared regulatory behaviour alongside factor-specific effects. Numerical suffix denotes isoform number.
We also see consistent structure at the level of transcription factor families. In Figure 6, members of the ETS and PITX families are enriched in overlapping regions of transcriptional space, suggesting that related factors can push cells towards similar broad states while still producing distinct local outcomes, as seen for example with ETV2_1 and ETV4_1. Biologically, this is exactly what would be expected if members of the same family share aspects of DNA-binding preference and regulatory logic, while retaining enough specificity to drive different downstream programmes. The emergence of this family-level pattern further supports the reproducibility of the screen and the consistency of opti-ox-driven programming, showing that the dataset is resolving meaningful biological structure across related factors.
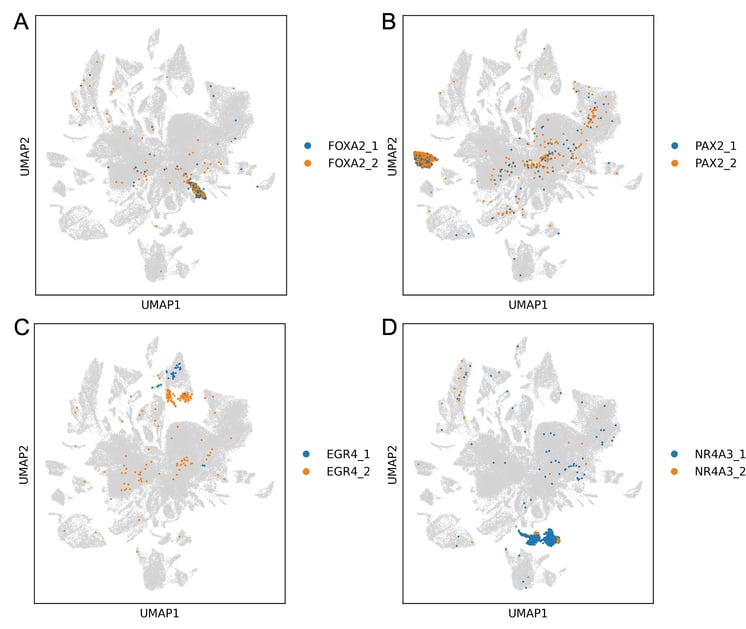
Figure 7. Isoform effects. Within clusters we can see isoform specific structure, though its variance is generally smaller than the average transcription factor effect. Numerical suffix denotes isoform number.
Designing this screen using opti-ox also allows us to examine transcription factor isoforms directly. Because the library encodes specific constructs, rather than relying on activation of the endogenous gene, we can ask whether different versions of the same factor produce similar or distinct outcomes, while also creating a framework for exploring broader questions in gene regulation and synthetic biology.
Across isoform pairs, we see a range of effects. Some isoforms occupy the same regions of transcriptional space and are enriched in the same clusters, suggesting broadly similar activity (Figure 7 A,B). Others occupy nearby but non-overlapping regions, pointing to more local differences in the cell states they produce (Figure 7 C,D). These examples suggest that isoform choice can meaningfully affect the expression levels of the transcription factors and therefore the perturbation outcome. This highlights an advantage of construct-based overexpression approaches over CRISPRa, because the exact expressed sequence can be directly controlled rather than being limited to activation of the endogenous locus.
Together, these results show that this screen can resolve multiple layers of transcription factor perturbation biology in a single experiment. Media conditions establish distinct baseline cell states, providing different contexts in which to test the same perturbations. Within those contexts, transcription factor overexpression drives additional structure, including reproducible shifts towards defined cell states and more variable or context-dependent responses. At finer resolution, isoforms of the same transcription factor can produce similar, subtly different outcomes.
In addition to simply asking which transcription factors have an effect, this dataset begins to show how those effects may be enhanced by the state of the cell, how consistently they are reproduced, and how each isoform is expressed. This provides a foundation for moving beyond single-factor ranking towards models that capture how transcription factor identity and biological context combine to shape cell fate.
More broadly, this is the kind of systematic perturbation data that predictive biology will require. If virtual cell models are to move beyond cataloguing observed cell states towards predicting how cells respond to defined inputs, they will need datasets generated with this level of scale, consistency and contextual structure. bit.bio is building towards a comprehensive perturbation atlas spanning human cell state and cell fate. These datasets include orthogonal perturbation approaches, such as CRISPR activation of transcription factors, alongside higher-complexity transcription factor screens designed to explore additive and synergistic effects. By expanding both the breadth and depth of causal perturbation data, this work is helping to reveal more complex molecular mechanisms, improve predictive models for engineering human biology, and provide a valuable ground-truth resource for AI-based approaches to predictive biology like virtual cell modelling.
Do you have any questions?
Get in touch with our team by filling this form.