08.10.2021 | Published by bit.bio

Thanks to cutting edge genetic engineering that will shape the future of research and medicine, you can start using reprogrammed human cells in your lab right now.
The data speaks for itself - we’ve already proven just how well this system works. What that means is reliable and rapid access to an endless supply of reproducible human cells. Right now, you can get hold of ioWild Type Cells and ioDisease Model Cells from bit.bio. But there’s more to come. With opti-ox technology, we plan to make every cell type of the human body accessible. From replacing animal models at the bench to a new generation of cell therapies, this is something that could really transform your research.
The need for better cell reprogramming
Before we dive into the future of cell reprogramming, we need to look back at what's happened before, what we can learn and how we can build on it.
Scientists have been using cell reprogramming for making induced pluripotent stem cells (iPSCs) from adult tissue for years – ever since Shinya Yamanaka and his lab did it with adult mouse and human cells in 2006/2007. 1,2 Although, interestingly, this work built on the long-forgotten reprogramming research of Davis, Weintraub and Lassar who converted mouse embryonic fibroblasts to myoblasts (i.e. muscle progenitors) back in 1987.3
Clearly, all parties were on to something because the notion of reprogramming cells to become a completely different type of cell won Shinya Yamanaka and John Gurdon the 2012 Nobel Prize for Medicine.
But while cell reprogramming has a great pedigree and the methods developed work, there were still problems to solve - yields were low, protocols were long and difficult to execute with consistent results. Early work activated a cell’s pluripotency by retroviral delivery of four transcription factors: OCT3/4, SOX2, KLF4, and c-MYC – collectively referred to as OSKM, known also as the Yamanaka factors. What you end up with are these unique stem cells you can differentiate into any other cell, assuming you know the right differentiation protocol that is.
In 2009 a team from Harvard showed that you can replace SOX2 and c-MYC, both genes associated with cancer, with a single molecule they called RepSox.4 But despite successive improvements like this, early reprogramming methods had potential safety issues and low efficiencies. In 2012, researchers from London ‘primed’ cells to enter a what they called a partial-iPSC (PiPSC) state before differentiating them directly into endothelial cells.5 Although this did remove tumor-formation risk, the reprogramming efficiency was around 34% only.
A leap forward in reprogramming methods
So, while the principles were sound, the process of cell reprogramming was some way off from optimal. But with so much potential in differentiation protocols, from basic research to medicine, the research community remained committed to taking a significant leap forward. And then along came forward programming to do just that.
Researchers found they could force cells to express specific transcription factors that work as ‘lineage-specific master regulators’, which made it much easier to generate somatic cell types, and they could do it in just weeks. Zhang and team could produce a homogeneous population of neurons from embryonic stem cells (ESCs) or iPSCs by overexpressing just neurogenin-2 (NGN2) or NeuroD1.6
Forward programming methods like these tend to rely on lentiviruses to insert a transgene encoding the transcription factor into the cell’s genome. The problem with the lentiviral approach is it’s a bit of a scatter-gun method: it inserts the transgene randomly throughout the genome. Also, and unsurprisingly, stem cells don’t like to be tampered with. So much so that they have mechanisms in place to combat rogue genomic material: gene silencing.
Transgenic sequences cue local epigenetic modifications of the cell genome, like methylation, that induce gene silencing by chromatin compaction and cut off the transcriptional machinery’s access to that rogue genomic material (i.e., your precious transgene!). So, when you use a lentiviral method, you can end up with variable gene expression if you’re lucky and no gene expression if you’re not.7
But what if you could directly place a transgene somewhere ‘safe’ and in a configuration that makes it almost out of sight? It just so happens that we can do that. Genomic safe harbours (GSHs) are regions within the genome that readily allow transgene expression and also minimize the risk of unwanted interactions (like gene silencing) by the host genome.8 The most commonly used GSH is the adeno-associated virus integration site 1 (AAVS1), a virus integration site that sits on chromosome 19.
And that’s exactly where opti-ox tech puts a sort of ‘plug and play’ system that forces overexpression of those lineage-specific master regulators we mentioned earlier – right into GSHs such as AAVS1 and others. Furthermore, special tricks in the design of the opti-ox cassette removed residual silencing risks by keeping the silencing machinery further away. All you need then is the transcription factor combination for a particular cell lineage to plug in!
Switching on cell identity
You can think of the GSH a bit like a USB port. Your transgene construct is the program you’re plugging into the genomic ‘hard drive’. If all goes well the program runs and transcription factors start the process of switching on a specific lineage.
But how do you get that program to run? Well, we created a ‘switch’ from the Tet-ON system that we can easily control. The Tet-ON system has a transcriptional activator that responds to doxycycline – this is called the reverse tetracycline transactivator (rtTA). The other part of the Tet-ON system is an inducible promoter regulated by rtTA (Tet-responsive element, or TRE) that drives expression of the transgene.
As we alluded to earlier, the GSH is ‘safe’ because putting something here – as opposed to randomly in the genome – means you don’t disrupt any important endogenous process, or switch on anything you shouldn’t (like oncogenes). And indeed, this method results in precise genetic engineering of the transgene and thus dramatically improves protocol reproducibility – both things you want in your reprogramming protocol!
With your transgene in place, present in every single starting cell, you add doxycycline, the system switches on and the cell begins to take on a new identity. Within just days, pluripotency markers drop off and lineage-specific markers are expressed.
Forward programming human iPSCs into neurons is a great example of this. Pawlowski et al started by making NGN2 human iPSCs with the opti-ox system – using zinc fingers (although we now use CRISPR/Cas9) to plug in the transgene for inducible NGN2 – and then treated the cells with doxycycline. After just four days, expression of the pluripotency genes NANOG and OCT4 dropped off almost completely and key neuronal markers like NGN2, VGLUT2 and FOXG4 among others were way up (Figure 1). By day seven, the cells were very clearly identifiable as neurons.9 That was fast, very homogeneous and felt like a real “EUREKA!” moment – and we don’t get enough of those in research! This method forms the basis of our rapidly maturing, highly defined ioGlutamatergic Neurons for research and drug discovery (Figure 2).
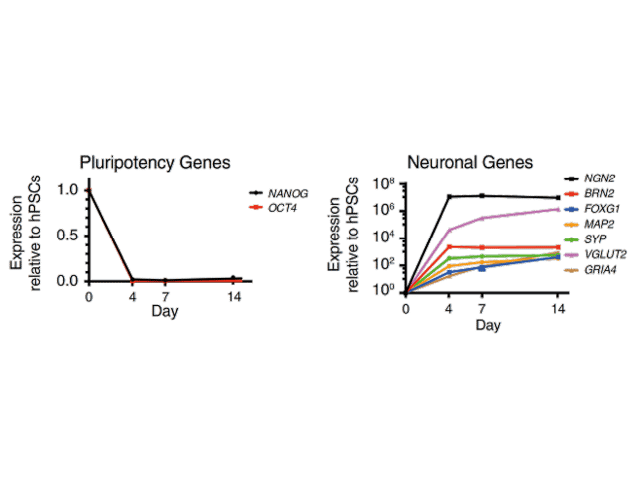
Figure 1. Neuronal Cell Identity a) Time course of neuron generation from hESCs by qPCR demonstrating the expression pattern of pluripotency factors (OCT4 and NANOG), pan-neuronal (MAP2 and SYP), forebrain (BRN2, FOXG1), and glutamatergic neuronal marker genes (VGLUT2, GRIA4) (n = 3 biological replicates; mean ± SEM; relative to PBGD and normalized to pluripotency). (Adapted from Figure 2 Pawlowski et al 2017)9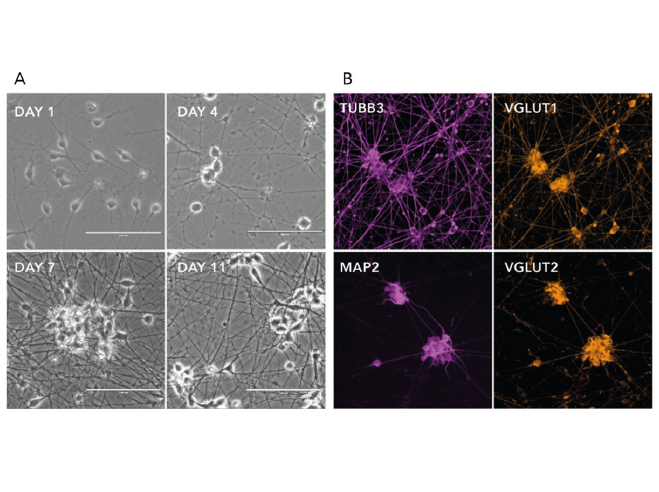
Figure 2. a) Phase-contrast images illustrating the morphological changes during neuron generation. b) Immunofluorescent staining on post-revival day 11 demonstrates homogenous expression of pan-neuronal proteins (MAP2 and TUBB3) and glutamatergic neuron-specific transporters (VGLUT1 and VGLUT2).
To test the system further, we went on to use the same approach but swapped out NGN2 for myogenic differentiation 1 (MYOD1), switched on opti-ox and this time successfully generated skeletal myocytes from human iPSCs with equivalent great speed and purity. We went on to develop the ioSkeletal Myocytes, characterised human myocytes for research and high-throughput screening focused on muscle, neuromuscular and associated metabolic diseases. We found that human stem cells, within days, convert into striated, multinucleated, and contractile skeletal myocytes, providing a scalable and consistent alternative to donor-derived and variable primary myocytes, and suboptimal animal models that do not accurately recapitulate human biology.
Now with knowledge of the correct transcription factor combination, we have the capability to generate an inexhaustible supply of fully reproducible human cells at a high yield too. This is a fast and scalable system that has the potential to really change how many of us conduct research.
A better model for testing
Where could we start using these cells right now? Well, for decades, we’ve relied on animal models to establish things like toxicity, efficacy, and target validation in the case of new drug development. But we all know that animal models can be expensive, are always time-consuming and, most importantly, aren’t always very relevant to human biology. That’s because animal models of disease are often wholly contrived.
Take Alzheimer’s disease. It’s true we know vastly more than before, about the accumulation of amyloid-β, which is derived from amyloid precursor protein (APP), and the involvement of tau protein. We even have mouse models that express high levels of APP in the brain and show a classic Alzheimer’s disease phenotype. But that phenotype is based on how we think Alzheimer’s disease works. Mice don’t develop Alzheimer’s naturally in the way humans do. As such, animal models like this just don’t reflect the true biology of the disease. This means the translational reliability and predictive value of a mouse model for something like Alzheimer’s disease therapeutics have been less than impressive.
So, if the preclinical work – before you get to the expensive clinical trials in humans part – is being carried out on flawed models, it’s no wonder that so many drug candidates fall down at later phases.10 Between efforts from organisations like NC3Rs11 to replace, reduce or refine animal use, and a growing body of evidence that the benefit of an animal model is outweighed by cumulative irreproducibility, harm and cost to humans,12 it’s become abundantly clear we need a better model.
On-demand, reproducible, human cells
And so, we come back to opti-ox-generated human cells, ioCells. You may be asking at this point, “But what about primary cells?”, which is a reasonable question. Primary cells obviously have great potential and more physiological relevance than an animal model. It’s just that they can be a pain to get hold of in sufficient quantities as they need to be isolated from humans. Primary cells also have consistency issues between batches – something that becomes especially troublesome once your research reaches a larger scale. There’s also the issue that some cell types are either inaccessible or only available in tiny quantities, such as cells from the human brain. Also, many primary cell types don’t proliferate much and so can’t easily be expanded.
ioCells sidestep those issues because they can be created at scale, with incredible consistency and efficiency and they won’t break the research budget either. All that’s required is the transcription factor combination of the desired somatic cell type. These combinations are already known for some cells like cardiomyocytes, neuron sub-types or muscle cells, but many more are still to be discovered to re-create the human body cellular diversity. The development of high-throughput screening methods for decoding cell identity regulators are on their way to identifying the minimal and sufficient genetic instructions to control cell fate… watch this space! Then, opti-ox will become your universal biological pen-drive to directly access an inexhaustible supply of reproducible human cell types, free of the ethical minefields around embryo and animal use but are loaded with the potential for research more relevant than ever.
We provide access to highly relevant human cells at scale unlike ever before, fulfilling the promise of the stem cell. The future of better, more relevant and more reliable models can be found in our ioCells.
References
- Takahashi, K. et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 131, 861–872 (2007).
- Takahashi, K. & Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126, 663–676 (2006).
- Davis, R. L., Weintraub, H. & Lassar, A. B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000 (1987).
- Ichida, J. K. et al. A Small-Molecule Inhibitor of Tgf-β Signaling Replaces Sox2 in Reprogramming by Inducing Nanog. Cell Stem Cell 5, 491–503 (2009).
- Margariti, A. et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue-engineered vessels. Proc. Natl. Acad. Sci. U. S. A. 109, 13793–13798 (2012).
- Zhang, Y. et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013).
- Smith, J. R. et al. Robust, Persistent Transgene Expression in Human Embryonic Stem Cells Is Achieved with AAVS1-Targeted Integration. Stem Cells 26, 496–504 (2008).
- Sadelain, M., Papapetrou, E. P. & Bushman, F. D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 12, 51–58 (2012).
- Pawlowski, M. et al. Inducible and Deterministic Forward Programming of Human Pluripotent Stem Cells into Neurons, Skeletal Myocytes, and Oligodendrocytes. Stem Cell Reports 8, 803–812 (2017).
- Harrison, R. K. Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 15, 817–818 (2016).
- National Centre for the Replacement Refinement & Reduction of Animals in Research.
- Akhtar, A. The Flaws and Human Harms of Animal Experimentation. Cambridge Q. Healthc. Ethics 24, 407–419 (2015).