18.01.2023 | Published by bit.bio
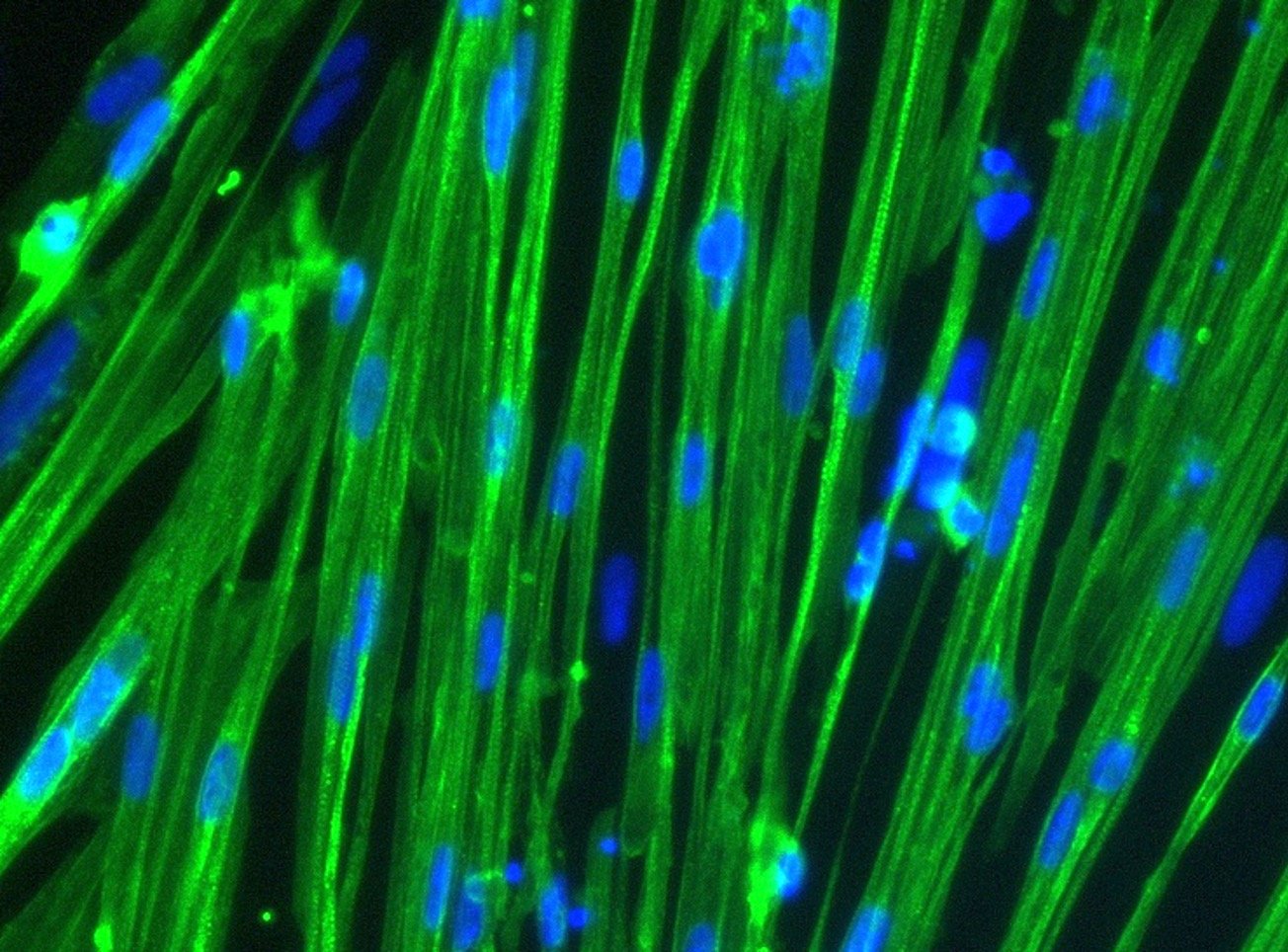
The human body is essentially a large assembly of muscle and water. Estimates suggest that approximately 40% of our mass is attributable to muscle. Not only do our muscles drive the mechanics of movement, but they also play a critical role in regulating energy metabolism as muscle cells (myocytes) consume and store large quantities of sugar. It’s no surprise then that muscle defects are prominent in many diseases.
Yet, despite its importance, muscle is a particularly challenging tissue to study, due in part to the difficulty of culturing myocytes. Like many mature cell types, myocytes do not regularly divide. This means access to large populations of differentiated myocytes is hard to come by.
The inability to faithfully model human myocytes in vitro has hindered progress in both understanding and treating musculoskeletal disorders. The World Health Organisation estimates that approximately 1.7 billion people currently live with a musculoskeletal disorder, and yet there are no known cures — treatment is limited to reducing symptoms and improving quality of life.
To facilitate research and improve patient care, researchers and drug developers need access to robust human myocytes on a large-scale. Fortunately, advances in human induced pluripotent stem cell (iPSC) technology have delivered exactly that.
In the sections that follow, we provide a brief overview of myocytes, how they’re currently modelled in vitro, and how tools for precision reprogramming of cell identity can advance the study of musculoskeletal disease.
What are skeletal myocytes?
Skeletal myocytes are terminally differentiated (mature) skeletal cells that represent the smallest building blocks in the larger muscle tissue hierarchy. As individual cells, skeletal myocytes express characteristic proteins such as actin, myosin, and dystrophin, all of which are critical for carrying out contractile functions. Skeletal myocytes fuse with one another to form multinucleated, filamentous structures known as muscle fibres which are then organised into higher-order bundles. Together, these bundles are capable of exerting considerable contractile force, enabling the body’s mechanical movement. When enough muscle fibres work together, the human body is capable of extraordinary feats, as demonstrated when Icelandic strongman Hafþor Björnsson set the deadlift world record of 501kg (1,104lbs)—more than 2.5 times his own body weight.
The mechanics of muscle contraction are well studied and described elsewhere. What’s important to know is that contraction requires ready access to large quantities of energy. Muscle tissue and specifically skeletal myocytes serve as a significant reservoir for sugar in the body, which can be stored as glycogen. These cells are also equipped with a high number of mitochondria to help convert this sugar into energy. Because of this, skeletal myocytes have an often under-appreciated role in the metabolic system.
In vivo, skeletal myocytes occupy an important niche in the body, facilitating both mechanical movement and sugar metabolism. Perhaps unsurprisingly, when genetic or environmental factors alter the activity of skeletal myocytes, the consequences can cascade to many different systems. Researchers are thus increasingly turning their attention to skeletal myocytes to better understand human physiology, both in health and disease. However, while abundant in the human body, accessing mature skeletal myocytes can be challenging.
Sources of skeletal myocytes
Researchers interested in studying human skeletal myocytes have few options. The most accessible cell types are immortalised cell lines which leverage tumorigenic mutations to drive continuous proliferation. However, as mentioned earlier, it is extremely unusual for myocytes to proliferate. While able to generate large quantities of cells, the artificial nature of these immortalised cells significantly reduces their resemblance to mature, non-malignant myocytes.
More human-relevant models can be found in patient-derived myocytes, which closely mimic in vivo phenotypes and, when sourced from individuals with musculoskeletal disease, can provide unique insight into cellular pathogenesis. However, these cell types are sensitive, challenging to culture, and cannot be expanded. As such, large-scale, statistically powered studies are often not feasible. Further, patient-to-patient variability can confound results.
To circumvent some of these problems, researchers have developed methods to create myocytes from human cells through transdifferentiation, wherein cells such as fibroblasts, are transformed into cells with a myocyte-like phenotype. Similarly, pluripotent stem cells can be subject to directed differentiation protocols in which time-sensitive exposure to growth factors is critical. While undoubtedly valuable, these approaches are both labour-intensive and generate impure populations of myocytes — neither of which is favourable for robust studies and large-scale drug screening.
Fortunately, developments in synthetic biology have opened the door to a new source of human myocytes through a process known as precision cellular reprogramming. In this new method, a specific collection of transcription factors is placed under the control of inducible elements and inserted into the genome of human iPSCs. This allows researchers to activate the genes that define cell fate in a controlled way, enabling highly efficient differentiation of the iPSCs into skeletal myocytes.
bit.bio uses these principles to generate iPSC-derived skeletal myocytes, and the high degree of efficiency offered by precision cellular reprogramming means that researchers can now access large quantities of mature human skeletal myocytes.
Dr Will Bernard from bit.bio discusses skeletal myocytes
Disease modelling with iPSC-derived human skeletal myocytes
Dr Rita Horvath, an academic neurologist at the Department of Clinical Neurosciences at the University of Cambridge, has been studying skeletal myocytes for over 25 years with a particular interest in learning how these cells contribute to mitochondrial diseases and other neuromuscular disorders.
For most of her career, Horvath’s in vitro studies have relied on primary myocytes collected directly from patient biopsies. From these samples, resident stem cells (so-called satellite cells) could be cultured and made to differentiate into more mature myocytes. With in vitro myocytes, Horvath and her peers have been able to closely study the mechanisms of disease pathogenesis and have begun to identify potential treatment modalities. However, it’s not been easy.
“There are some difficulties when we want to study muscle cells for drug development,” Horvath explained. “If we really want to study the muscle cells that are closest to skeletal muscle cells, we differentiate satellite cells into myotubes which can only live for about 3 weeks. So, we have to keep differentiating new muscle cells every time.”
This is problematic for many reasons, not least of which being that primary muscle cells have been harder to come by in recent years (due in part to the use of next generation sequencing technology for disease profiling, rather than tissue biopsies). As a result, each experiment is precious, and each study must be laser focused — a reality that greatly limits the pace of progress.
For Horvath, access to a stable, scalable population of human myocytes is an exciting prospect because it means they don’t have to be so selective about what they look at in each experiment. With a large population of cells, she says, “we can study the activity of enzymes, or the amount of different proteins, or amino acids. And this may enable us to identify potential options for treatments.”
Dr Rita Horvath describes how myocytes play a role in various diseases
In muscular dystrophies such as Duchenne muscular dystrophy, faulty sarcolemma proteins are responsible for membrane defects that lead to debilitating muscular defects. Modelling such a disease has been challenging. In vivo models have been made, however significant differences in physiology and genetics limit their human relevance. According to Luke Flatt, a former Senior Scientist at Charles River Laboratories, mouse models of Duchenne muscular dystrophy (DMD) display considerable muscle regeneration and displacement in genetic models, neither of which is observed in humans.
Flatt’s research has focused on establishing models of various musculoskeletal disorders. In a recent webinar, Flatt spoke about his experience with these models and the need for better, more human-relevant skeletal myocytes. You can listen to his presentation here.
With patient derived cells in short supply and a lack of robust in vivo models, iPSC technology is proving invaluable. In addition to providing a stable source of healthy myocytes, gene editing can be used to introduce disease-specific mutations, such as a DMD-relevant exon deletion. Pairing an unedited “wild type” cell with a disease model allows researchers to directly attribute variation in their data to the disease causing mutations. Such precision is powerful as it supports the confident scale-up of experiments from single assays to multiplexed read-outs, increasing the throughput by which candidate therapeutics can be tested for their efficacy and toxicity.
Whether for disease modelling, drug screening, or both, having access to large, robust stocks of human myocytes is critical. bit.bio’s human iPSC-derived skeletal myocytes overcome boundaries that have limited the field for decades, enabling more human-relevant modelling of a tissue type that is so prominent and yet so often overlooked.
References
- Spendiff S, Reza M, Murphy JL, Gorman G, Blakely EL, Taylor RW, Horvath R, Campbell G, Newman J, Lochmüller H, Turnbull DM (2013) Mitochondrial DNA deletions in muscle satellite cells: implications for therapies. Hum Mol Genet. 2013 Dec 1;22(23):4739-47. doi: 10.1093/hmg/ddt327. Epub 2013 Jul 11. PMID: 23847047; PMCID: PMC3820134.
- Olimpio C, Tiet MY, Horvath R (2021) Primary mitochondrial myopathies in childhood. Neuromuscul Disord. 2021 Oct;31(10):978-987. doi: 10.1016/j.nmd.2021.08.005. PMID: 34736635.
- Hathazi D, Griffin H, Jennings MJ, Giunta M, Powell C, Pearce SF, Munro B, Wei W, Boczonadi V, Poulton J, Pyle A, Calabrese C, Gomez-Duran A, Schara U, Pitceathly RDS, Hanna MG, Joost K, Cotta A, Paim JF, Navarro MM, Duff J, Mattman A, Chapman K, Servidei S, Della Marina A, Uusimaa J, Roos A, Mootha V, Hirano M, Tulinius M, Giri M, Hoffmann EP, Lochmüller H, DiMauro S, Minczuk M, Chinnery PF, Müller JS, Horvath R (2020) Metabolic shift underlies recovery in reversible infantile respiratory chain deficiency. EMBO J. 2020 Dec 1;39(23):e105364. doi: 10.15252/embj.2020105364. Epub 2020 Oct 31. PMID: 33128823; PMCID: PMC7705457.
- Thompson R, Spendiff S, Roos A, Bourque PR, Warman Chardon J, Kirschner J, Horvath R, Lochmüller H (2020) Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol. 2020 Jun;19(6):522-532. doi: 10.1016/S1474-4422(20)30028-4. Epub 2020 May 26. PMID: 32470424.