07.09.2023 | Published by bit.bio
In the early 1800s, smattered reports began to circulate in the medical literature describing a mysterious disease with perplexing symptoms. At first glance, the patients appeared to have bulging musculature with notably well-defined and rigid muscles that some physicians likened to statues of Hercules1. That this physique was seen in children no more than 14 years old was alarming, but even more so was the paradoxical weakness they all suffered. Whatever the disease was, it appeared to wreak havoc on skeletal and cardiac muscles, robbing patients of both mobility and life beyond adolescence.
It’s now believed that these early reports described patients with Duchenne Muscular Dystrophy (DMD), a particularly severe form of muscular deterioration wherein a patient’s muscle tissue is unable to withstand the stress of routine use. The firm and well-sculpted muscles that beguiled physicians of the 19th century were little more than the product of the patients’ bodies attempting to repair damaged muscle. As described by Guillaume Benjamin Amand Duchenne, the eponymous French neurologist, “these muscle contours, in appearance so formidable, are formed principally by muscle fibres stuffed with interstitial connective tissue and interstitial fibrosis1.”
Today, DMD is known to be a genetic disease affecting families around the world. According to a recent meta-analysis involving more than 40 demographic studies, approximately 2.8 out of every 100,000 people globally are affected2. Advances in medical care have nearly doubled the average DMD patient’s lifespan, but a cure remains elusive3. Fortunately, recent advances in molecular biology have shed light on the pathological mechanisms of DMD and have inspired the development of a promising new therapeutic approach involving antisense oligonucleotides (ASOs).
Below, we delve into the molecular pathology of DMD and how new developments in ASO technology and exon-skipping therapy may improve patient outcomes.
What is Duchenne Muscular Dystrophy (DMD)?
Duchenne muscular dystrophy (DMD) is a fatal form of muscular dystrophy caused by mutations in the dystrophin gene that decrease myocyte integrity, ultimately leading to a progressive degeneration of skeletal and cardiac muscle tissue3.
In healthy myocytes, the dystrophin gene produces a protein by the same name (dystrophin), which has critical functions in shock absorption during muscle contraction and relaxation. Specifically, the Dp427m isoform of dystrophin is an essential structural protein in what’s known as the dystrophin-associated protein complex, or DAPC—a large and varied assemblage of proteins that connect key cytoskeletal proteins (such as F-actin) to both the extracellular matrix (ECM) and the sarcolemma (the muscle fibre’s plasma membrane)3,4. This connection allows for the mechanical stress generated by repeated cycles of muscle contraction to be dissipated in the same way that shocks on a car will protect it from potholes.
The Inheritance of DMD
DMD is an X-linked recessive disorder, meaning individuals with at least one functioning copy of the dystrophin gene are unlikely to develop DMD3. Therefore, a person's odds of developing the disease primarily depend on the number of X chromosomes—and by extension, dystrophin gene copies—they inherit. Accordingly, DMD is most common among individuals who inherit only one X chromosome. Within this population, approximately 19.8 per 100,000 live births develop DMD2.
Mutations that disrupt dystrophin’s ability to interact with the cellular cytoskeleton, sarcolemma, or ECM greatly reduce the myocyte’s ability to handle stress and, as a result, leave it vulnerable to damage during routine use. Such is the case in DMD.
Thousands of mutations in the dystrophin gene have been linked to DMD, most of which disrupt the dystrophin gene’s reading frame, resulting in a truncated, non-functional protein5. With low expression levels (typically <10%) and severely compromised structure, the aberrant dystrophin is unable to fill its critical functions. This lack of functional dystrophin disrupts cytoskeletal and focal adhesion structures, loss of myocyte resilience under contractile stress, functional ischemia, tissue necrosis, inflammation, and fibrosis, among many other effects3,4,6.
Clinically, this manifests as progressive weakness and muscle degeneration, first in the distal muscles that normally express high levels of dystrophin (like the quadriceps) and later in chronically active muscles, such as the diaphragm3,4. Efforts to prevent this degeneration have thus far been unsuccessful, with patient care primarily focused on managing inflammation and reducing muscle stress. However, a growing understanding of the genetics behind DMD and a closely related condition known as Becker muscular dystrophy (BMD) has fueled hopes for more curative approaches, ones that restore dystrophin’s reading frame and, in turn, the patient's ability to produce functional dystrophin protein.
Duchenne vs. Becker Muscular Dystrophy
To understand modern therapeutic development for DMD, it helps first to discuss BMD. Like DMD, BMD is caused by mutations in the dystrophin gene that lead to skeletal and cardiac muscle deterioration. Unlike DMD, though, patients with BMD may only experience degenerative effects in their later teenage years, with some patients only ever showing very limited dystrophy. Such a substantial difference between two diseases born from mutations in the same gene is attributed to BMD patients harbouring mutations that preserve the dystrophin gene’s reading frame, even while producing proteins with large internal truncations4,7,8.
Though less protective against muscle damage when compared to the full-length dystrophin protein, extensive research into the genetics of BMD and DMD suggests that truncated versions of the dystrophin protein can help stave off severe muscular dystrophy so long as they are stable and maintain a partial ability to engage in critical protein-protein interactions (such as those with F-actin)4,8. Thus, mutations that cause DMD lead to severe muscular dystrophy because out-of-frame mutations generate significant truncations that eliminate key protein-protein binding domains and produce instability.
This observation does more than explain the difference between DMD and BMD; it also introduces a potential therapeutic strategy: If researchers can find a way to restore the dystrophin gene’s reading frame, they may be able to prevent the complete loss of protein function and subsequently severe muscular deterioration that’s typical of DMD9.
To do this, scientists have turned to antisense oligonucleotides and the “exon skipping” strategy.
Exon Skipping Therapy and Antisense Oligonucleotides
Antisense Oligonucleotides (ASOs) are modified strands of nucleotides about 15-20 bases long designed to alter the processing of specific RNA transcripts, thereby influencing the expression of specific proteins10. With respect to DMD, ASOs are being developed to alter the splicing of the dystrophin RNA so that it restores the reading frame at the cost of internal truncation (Figure 1). This approach would effectively transition patients from a DMD phenotype to the more mild BMD phenotype, leading to a better prognosis and quality of life9.
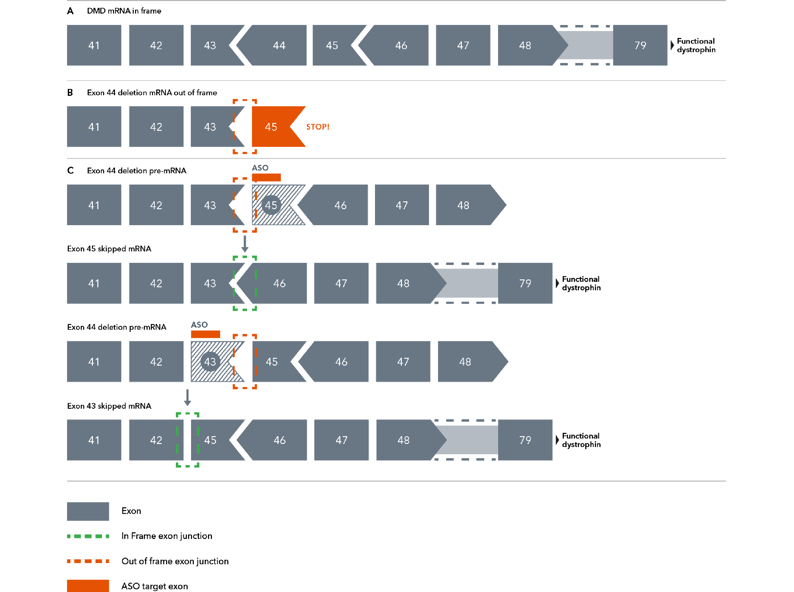
Figure 1. Diagram demonstrating the theory behind exon skipping therapy. Chevrons between exons (grey boxes) represent positions where codons span exon-exon junctions. Here, a deletion of one such exon (exon 44) leads to an out-of-frame mutation due to a now incomplete codon. Antisense oligonucleotides can manipulate the spliceosome into restoring the reading frame by skipping over exon 43 or exon 45.
Briefly, the dystrophin gene is the largest known protein coding sequence in the human genome, with codons describing the protein’s >3,000 amino acids distributed across 79 exons4. Some of those codons span exon-exon junctions, meaning the codon begins on one exon and terminates on another. Should a mutation disrupt these junction-spanning codons, such as the deletion of an exon containing an incomplete codon, the gene’s open reading frame can be thrown off, resulting in a premature stop codon and a critically shortened protein (Figure 1)11.
Approximately 65% of DMD-causing mutations are exonic deletions that do exactly that, altering the gene’s reading frame with disastrous effects. Another 20% of patients harbour either nonsense mutations (10% of all patients), small indels (7% of all patients), or splice site mutations (3% of all patients) that collectively lead to the same result: a severely truncated and non-functional dystrophin protein5,9.
The deletion of exon 44 interrupts a junction-spanning codon, meaning there is now an incomplete codon (sometimes known as an “out-of-frame” codon) at the end of exon 43. Suppose exons 43 and 45 are spliced together. In that case, the incomplete codon will shift the reading frame, resulting in the rest of the dystrophin protein not being transcribed correctly, leading to the non-functional truncation. ASOs solve this codon problem in a few ways (Figure 1C)10,11:
- ASOs can direct the cell to exclude exon 43 altogether, allowing exon 42 and 45 to generate a complete reading frame, leading to a truncated but functional dystrophin protein or
- ASOs can direct the cell to exclude exon 45, which shares a junction-spanning codon with its neighbour, exon 46. Because of this, the loss of exon 45 would create an incomplete codon on exon 46. When the incomplete codon on exon 43 is spliced together with the incomplete codon on exon 46, they create a complete codon that restores the reading frame.
iPSC Derived Skeletal Muscle Cells for DMD Research
bit.bio has developed human iPSC-derived skeletal myocytes harbouring a deletion of exon 44 or exon 52 in the dystrophin gene. The cells show a lack of dystrophin by immunocytochemistry, and dystrophin restoration has been demonstrated at the mRNA and protein levels by ASO exon skipping. These cells offer a rapidly maturing, consistent, and scalable isogenic system to study Duchenne muscular dystrophy in a physiologically relevant human cell model.
Learn more about ioSkeletal Myocytes for researching DMD >
In recent years, four ASO-based therapeutics have received FDA approval for the treatment of DMD, each following an exon skipping strategy12. All ASO-based therapeutics for DMD aim to manipulate splicing machinery so that it skips over key mutational hotspots in the dystrophin RNA, particularly regions between exons 45 to 55 (wherein approximately 70% of the exon-deletions that cause DMD are clustered5).
The need for a scalable model of DMD
While ASOs are promising, their development is not without significant challenges. Because ASOs target RNA and not the underlying genomic mutations, therapeutic benefit is dependent on successfully delivering and maintaining high enough concentrations of ASOs in target tissues. Such issues are not trivial in the dynamic in vivo environment, and this may need to be done throughout the patient’s lifespan. Therefore, ASOs must be safe, highly specific, and have favourable pharmacokinetics9,10,13.
These issues are familiar to the drug development industry, which has learned to use high-throughput screening and an iterative design-build-test-learn cycle to optimise small molecules. However, doing so with ASOs will require scalable and robust models of DMD that closely replicate human disease mechanics. Fortunately, advances in induced pluripotent stem cell (iPSC) technology have enabled the creation of iPSC-derived skeletal myocytes containing DMD-causing mutations. With bit.bio’s opti-ox technology, these cells can be rapidly produced on a large scale, enabling high-throughput testing of ASOs and ultimately helping to accelerate ASO development.
Now, nearly 200 years after the first medical descriptions of DMD, exon skipping therapy brings us significantly closer to treating this destructive disease.
References
- Tyler, Kenneth L. “Origins and Early Descriptions of “Duchenne Muscular Dystrophy.”” Muscle & Nerve, vol. 28, no. 4, 2003, pp. 402–22.
- Crisafulli, Salvatore, et al. “Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis.” Orphanet Journal of Rare Diseases, vol. 15, no. 1, 5 June 2020.
- Duan, Dongsheng, et al. “Duchenne Muscular Dystrophy.” Nature Reviews Disease Primers, vol. 7, no. 1, 18 Feb. 2021, pp. 1–19.
- Gao, Quan Q., and Elizabeth M. McNally. “The Dystrophin Complex: Structure, Function, and Implications for Therapy.” Comprehensive Physiology, vol. 5, no. 3, 24 June 2015.
- Aartsma-Rus, Annemieke, et al. “Theoretic Applicability of Antisense-Mediated Exon Skipping for Duchenne Muscular Dystrophy Mutations.” Human Mutation, vol. 30, no. 3, Mar. 2009, pp. 293–299.
- Wilson, Darren Graham Samuel, et al. “The Role of the Dystrophin Glycoprotein Complex in Muscle Cell Mechanotransduction.” Communications Biology, vol. 5, no. 1, 27 Sept. 2022.
- Mercuri, Eugenio, et al. “Muscular Dystrophies.” The Lancet, vol. 394, no. 10213, Nov. 2019, pp. 2025–2038.
- Monaco, Anthony P., et al. “An Explanation for the Phenotypic Differences between Patients Bearing Partial Deletions of the DMD Locus.” Genomics, vol. 2, no. 1, Jan. 1988, pp. 90–95.
- Niks, Erik H., and Annemieke Aartsma-Rus. “Exon Skipping: A First in Class Strategy for Duchenne Muscular Dystrophy.” Expert Opinion on Biological Therapy, vol. 17, no. 2, 23 Dec. 2016, pp. 225–236.
- Dhuri, Karishma, et al. “Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development.” Journal of Clinical Medicine, vol. 9, no. 6, 26 June 2020, p. 2004.
- Kole, Ryszard, and Arthur M. Krieg. “Exon Skipping Therapy for Duchenne Muscular Dystrophy.” Advanced Drug Delivery Reviews, vol. 87, June 2015, pp. 104–107.
- Egli, Martin, and Muthiah Manoharan. “Chemistry, Structure and Function of Approved Oligonucleotide Therapeutics.” Nucleic Acids Research, vol. 51, no. 6, 7 Mar. 2023, pp. 2529–2573.
- Migliorati, Julia M., et al. “Absorption, Distribution, Metabolism, and Excretion of US Food and Drug Administration–Approved Antisense Oligonucleotide Drugs.” Drug Metabolism and Disposition, vol. 50, no. 6, 1 June 2022, pp. 888–897.